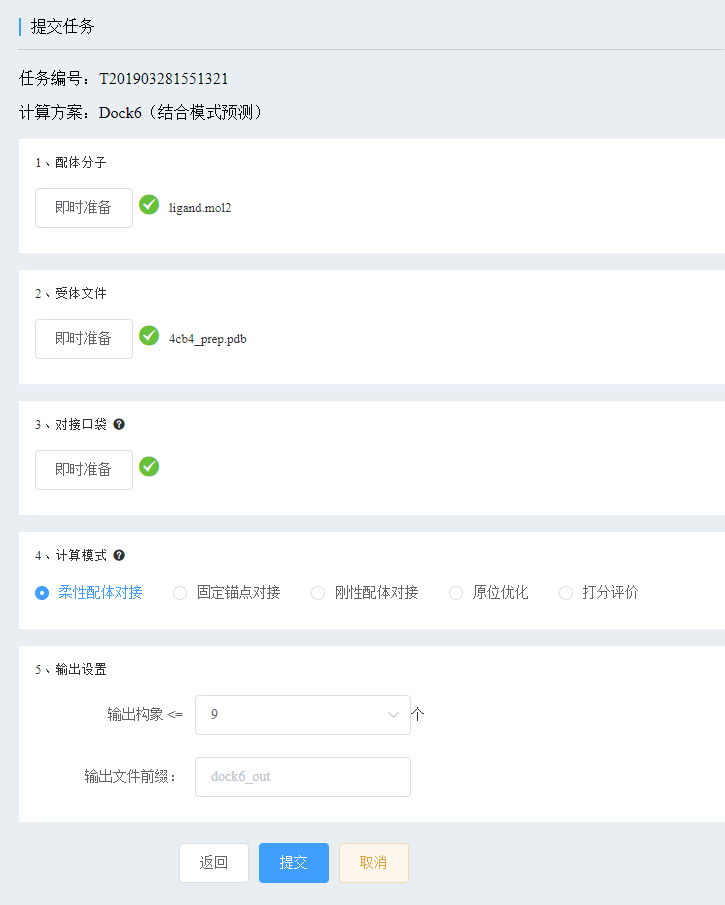
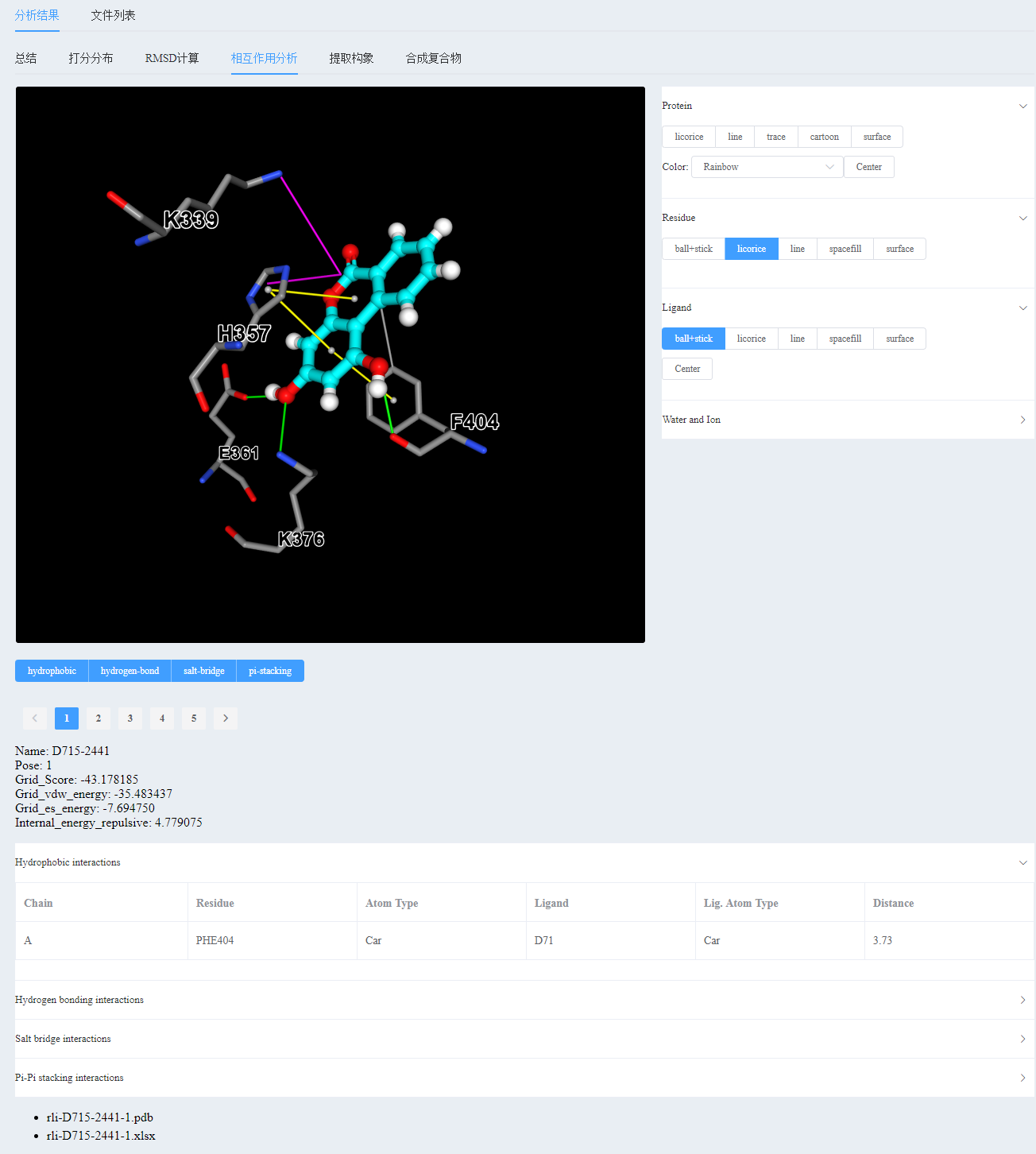
From this issue, Xiaobian will lead you to use the Yin Fu cloud computing platform (http://cloud.yinfotek.com/) to do a series of calculations "articles". In this issue, we will reproduce the calculation results of a 2018 document [1] to see how the author uses molecular docking techniques to clarify the mechanism and sublimate the article. The literature has an impact factor of 4.6 , which is a typical “ experiment + calculation †model, which is close to the research status of most researchers. As long as you are familiar with this model, you can publish the same level of paper with a little effort. The authors found a novel anti-influenza virus compound D715-2441 from more than 8,000 small molecules by cellular MTT assay. Further experiments have found that it has antiviral activity against various subtypes of influenza A virus (H1N1, H5N1, H7N9, H3N2, clinical isolate 690 (H3) and oseltamivir resistant strain with H274Y NA variation). Inhibits the early stages of viral replication. More importantly, D715-2441 significantly inhibited the activity of viral polymerase and directly affected the localization of PB2 protein. Binding affinity analysis confirmed that the compound specifically binds to the PB2 cap protein. In order to elucidate the mechanism of action of inhibitor D715-2441, the authors used DOCK 6 molecular docking technology to analyze its binding mode. In simple terms, the calculation steps for molecular docking consist of 4 steps: 1. Prepare ligands (small molecules) and receptors (biomacromolecules, such as proteins) structures; 2. Define the interface bag; 3. Perform docking calculations; 4. Analyze the docking results (scoring and combining mode analysis). Below, Xiaobian textually teaches you to use DOCK6 scheme of Yinfu cloud computing platform to complete docking calculation and analysis. This tutorial is not intended to cover every step of the process in detail, but to clarify the general process and key points. But there are a lot of notable details, which is a general experience that you can't see on the tutorial. If the reader is not very familiar with the operation of this platform, please refer to the corresponding tutorial ( Molecular Docking Combined Mode Prediction Tutorial ). Molecular docking calculations require the preparation of three-dimensional structures of ligand small molecules and receptor proteins. The ligand structure is usually small and simple, and can be constructed by software, but the protein structure is large and complex, and the experimentally resolved structure is usually used, which can be downloaded from the RCSB protein database. The PB2cap protein PDB number used in this work is 4CB4. Please download it at https:// (4cb4.pdb file). Log in to the platform, create a project, and fill in the content. “Simplified Remarks†and “Detailed Description†are for you to see, but you can leave it blank, but filling out the memo is a good habit to encourage. Then, choose a calculation plan or tool. Here, we choose the Dock6 (combined pattern prediction) scheme of the same paragraph as the literature. After that, fill in the task content . The platform provides a variety of input methods for preparing ligand molecules, the simplest of which is the online drawing structure (draw small molecules). Draw the chemical structure of D715-2441 on the artboard and fill in the molecular name ( do not have Chinese, spaces, special symbols or other languages ​​except English letters! ) and click the Add button. The input molecules need to be processed for docking, including: adding hydrogen atoms, adding charge, generating 3D structures, and minimizing energy. The platform has been optimized for most situations, so the default settings are fine. It is worth noting that: 1. MMFF94 force field is one of the best force fields for organic small molecules, including some metal elements [2], but not all elements of the chemical periodic table are supported. If necessary, please use UFF force. Field; 2. The force field used for charge and energy optimization is preferably the same. Click the Ready button. We see the processed ligand structure from the view. Although the platform has been done very well, manual inspection is still necessary. Please check carefully whether your structure is correct according to your chemistry knowledge, such as whether the price key is balanced, whether the structure is not twisted, and whether the configuration is correct. Once you have no problem, click the OK button; otherwise, go back to the front, repaint the structure or use another input method. Unlike ligands, receptor molecules are usually biological macromolecules that cannot be prepared by hand. The platform provides additional input methods. Click Upload File, select the 4cb4.pdb file you just downloaded, and follow the prompts. In general, the crystal structure file downloaded from the PDB library contains many components, not only (single or multiple) peptide chains, but also organic small molecules and solvent molecules (water molecules, metal ions). If the NMR structure is downloaded, it may also contain multiple protein conformations (called models). In the step of preparing the receptor structure, we need to define which model to use (default is 1), which part of the structure is classified as a receptor, which part is a ligand (which will be extracted for pocket positioning; if not, it may not be defined ) and Other impurities. Here, we choose A:PROTEIN as the receptor, A:MGT1484 as the ligand, and the unselected part will be deleted by the program in the subsequent processing. We need to define the approximate area of ​​the compound D715-2441 combination - the interface pocket. In the above steps, we extracted the ligand molecules originally present in the crystal structure. It is known from the literature that its binding position is the possible binding area of ​​D715-2441. In fact, for this protein, it is only here. Select MGT.pdb from the list of pop-up files by selecting the file method and click on the display box . Wait a moment, a green box will appear in the view to surround the MGT molecule. What we need to ensure is that the box is large enough to enclose the pocket. The default setting is basically sufficient, and if necessary, increase the sphere range (which may cause the box to expand). Then click to generate a grid point . This step is usually a long time, please be patient. Then click the OK button. The Dock6 program provides us with a variety of docking calculation modes, which are explained slightly here. In various modes, the protein receptors are rigid and immobile. Because Dock6's Grid algorithm does not actually use the protein structure for docking, but uses its model (which is the lattice point generated earlier). According to the way of ligand movement, it is divided into the following cases: 1) Flexible ligand docking , ie, the ligand is flexible in the docking process, and the single and double bonds can be rotated to produce different conformations. This is the most common default mode; 2) Fixed anchor point docking , that is, a small-scale conformational search of the ligand in the pocket. The ligand has a structure (usually the largest rigid structure) that appears to be fixed while the other parts are variable. The use of this mode is to find a better combination mode while keeping the overall binding position of the ligand unchanged. 3) The rigid ligands are docked , that is, the ligand is rigid, and the chemical bonds cannot be rotated, and only the whole can be translated and inverted. A possible use scenario is that the conformation of the ligand has a special meaning, it is desirable to remain unchanged after docking, or to assess whether the protein can accommodate this conformation; 4) in situ optimization (in-situ minimization), i.e. the energy of the ligand molecule in the pocket optimization, conformational search is not performed. Can be used to optimize the binding mode of the ligand; 5) Score evaluation , that is, the score of the ligand molecules in the pocket is evaluated, and the conformation search is not performed. A different scoring function can be used to re-score a certain binding mode of the ligand. We use the flexible ligand docking mode, the other parameters take the default values, and then submit the task. The docking total score of the Dock6 scheme is Grid Score. Negative values ​​indicate that there is a combination, and positive values ​​indicate no binding. Therefore, the smaller the score value (the larger the absolute value of the negative value), the stronger the binding force. It is related to two other energy terms - van der Waals force and electrostatic force - Grid Score = Grid_vdw + Grid_es. Internal Energy is the intramolecular energy, which indicates the tension of the butt joint. The positive value is as small as possible. Usually 20 kcal/mol or less is reasonable. The Pose column is the docking constellation number, arranged from small to large according to the Grid Score. Therefore, the first pose is the docking constellation that the docking software judges to be the best based on the scoring. According to experience, Grid Score>-40 kcal/mol is a poor binding force, -40~-50kcal/mol is moderate, and <-50kcal/mol is better. Since the D715-2441 molecule is small, the hydrophobic interaction area is small, the van der Waals force contribution is naturally small, and the electrostatic force contribution is usually not large, resulting in poor overall score. This is understandable, and it should be understood that the docking conformation is reasonable. From the distribution of scoring, the scores of each pose are not much different, and Van der Waals forces contribute. The electrostatic contribution of the first pose is surprisingly large compared to other poses, meaning that it has more strong electrostatic forces (such as hydrogen bonds). And salt bridge). Look closely at each pose and combine the above docking scores. We believe that the first pose has the most force, the best combination, and the combination is the best conformation. In contrast to the literature, this pose is almost identical (the differences are explained below). Therefore, we chose it to do the combined mode analysis. For the sake of clarity, click on the cartoon button under the Protein on the right side of the view to remove the cartoon style. Click on the center button under Ligand to center the ligand. We clearly see that the aromatic ring structure of compound D715-2441 forms a sandwich structure between the imidazole ring of H357 and the benzene ring of F404, and forms a Ï€-Ï€ stacking effect (yellow line) with F404, respectively. The hydrophobic interaction (grey line), these forces together constitute the van der Waals force, providing a binding force contribution of about -35.48 kcal/mol. On the other hand, the carboxyl group of E361 and the protonated N atom of K376 simultaneously form a hydrogen bond with the hydroxyl group on the benzene ring , and the hydrogen bond distance and angle can be found from the table below. The oxygen atom on the F404 backbone also forms a hydrogen bond with another hydroxyl group of D715-2441 . Unlike the literature, both the N+ atom of K339 and the imidazolyl group of H357 form a salt bridge with the cyclic ester group . This effect is generally considered to be atypical, so it is not mentioned in the literature, but it is presented in our detection and analysis tools. These polar effects provide a strong electrostatic force contribution (about -7.69 kal/mol) for the combination of the compounds, which is why the electrostatic force is very significant compared to other poses . Another difference from the literature is that the literature shows that Phe323 has a Ï€-Ï€ stacking effect with the compound, and Phe325 has a hydrophobic effect. But in our analysis, these two forces did not appear. In fact, these two amino acid residues are a little farther from the substrate and just exceed the analytical criteria for the interaction force of the analytical tool. However, if the atomic structure is displayed, it is found to be nearby (below) . In the flexible ligand docking mode, the protein is rigid, and the shape and residue position of the protein pocket are not necessarily the closest to the ligand. It is generally believed that there is an "inducing fit" effect between protein-ligands. In the real situation, due to molecular thermal motion, the distance between protein and ligand cannot be fixed. The docking calculation only shows its static side. In the real world, the force of the geometric distance near the critical point may be “loomingâ€. High quality articles require high quality Figures. Xiaobian will teach you to use PyMOL as a superior protein-ligand binding pattern in the next issue. Please pay attention~ [1] Teng et al. A Small-Molecule Compound Has Anti-influenza A Virus Activity by Acting as a “PB2 Inhibitorâ€. Mol. Pharmaceutics, 2018, 15 (9), pp 4110-4120. DOI: 10.1021/acs. Molpharmaceut.8b00531 [2] https://open-babel.readthedocs.io/en/latest/Forcefields/mmff94.html#mmff94-force-field Medical Ear Thermometer,Ear Thermometer,Infrared Ear Thermometer Dongguan Keyutai Mask Co., Ltd. , https://www.maskkytai.com
2, create projects and tasks 






1) View the docking score 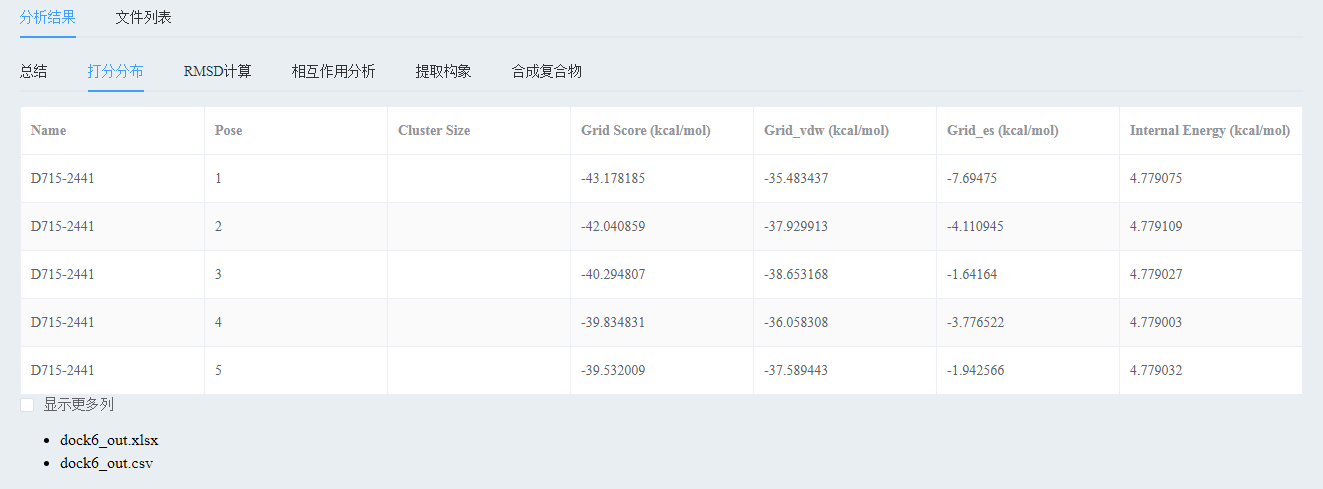

[Reproduction of the literature] Molecular docking technology to study the binding mode of D715-2441 inhibitor to PB2 protein
[Foreword]
ã€Research Background】
ã€calculation steps】
1. Preparation for the preliminary work
3. Prepare ligand molecules
4, prepare the receptor molecule
5, define the interface bag
6, submit a computing task
7, analysis of docking results
2) Analyze the interaction
[Next notice]
ã€references】